
Author: Silas Minnick, Senior Quality Engineering Consultant
Are you preventing risk or vulnerable to it? Whether you are a medical device manufacturer or a quality professional, this guide will help you master the risk management process. Drawing on the latest updates and changes in industry guidelines, including ISO/TR 24971:2020 and ISO 14971:2019, we will provide you with expert insights and practical strategies to identify hazards, evaluate risks and develop mitigation plans. Let’s dive into the world of medical device risk management and unlock the key to successful medical device development.
Guidance for Medical Device Risk Management Process
In addition to reducing foreseeable hazards, risk management plans help organizations comply with healthcare regulations and standards issued by governing bodies such as the FDA or ISO. The risk management guidance for medical devices, ISO/TR 24971:2020, was released in June 2020. It defines the terminology, principles, and processes for applying risk management to medical devices.
In December 2019, the latest revision to ISO 14971 was released (ISO 14971:2019). This standard specifies the terminology, principles, and processes related to applying risk management to medical devices. For European manufacturers, BS EN ISO 1497:2019+A11:2021 is the directive.
Medical device companies are empowered to determine the risk analysis process for any associated risks and determine protective measures to be taken when necessary.
What is the best ISO 14971 risk management process?
The risk management process should be conducted in three steps:
- Step 1: Identifying hazards
- Step 2: Risk evaluation to determine the severity of the risk
- Step 3: Manage risk with a mitigation plan
Step 1: Identifying Hazards
The first step in a risk management plan is to identify potential hazards. This involves analyzing the environment, processes, and people affected to identify all foreseeable risks. Once identified, the risks can be assessed according to their severity and probability of occurring. A good risk management plan should consider both physical and human-induced hazards so that you can implement appropriate controls to reduce or eliminate potential harm.
According to the ISO/TR 24971:2020, a medical device should have a positive impact or desirable outcome on the health of an individual, patient management, or public health. These benefits come from the intended use of a medical device, and risk management is necessary to ensure that misuse is mitigated. This includes using a product or system in a way not intended by the manufacturer and can result from readily predictable human behavior, intentional or unintentional.
Some common hazards to look out for include the following:
- Cybersecurity hazards. Medical device manufacturers must remain vigilant and take proactive measures to prepare for potential cybersecurity attacks. The widespread adoption of digital health technologies has made the medical device industry an increasingly attractive target for cybersecurity hackers, and risk management in this area is crucial.
- Physical hazards, including fire, electric shock, or radiation.
- Mechanical hazards, such as sharp edges on components.
- Human-induced hazards should be considered in the risk management process, which may include intentional misuse of a medical device.
For example, a newly created pacemaker could have several identified hazards, including biohazard contamination before implanting the pacemaker, caused by lack of proper sterilization, leading to infection in a patient. Another possible and easily identifiable hazard could be an electrical failure of the power source, leading to malfunction.
Check out these seven must-haves for a medical device quality system.
Step 2: Risk Evaluation to Determine the Severity of the Risk
Immediately after you identify hazards, you must evaluate the hazard with risk acceptability criteria. There are two aspects to consider for the criteria: the severity of a hazard and the probability of it occurring.
Consider these criteria and establish zones to categorize foreseeable hazards. Here we outline three potential zones:
- Low Zone: Hazards with minimal severity and infrequent probability of occurring are considered acceptable, and any mitigation measures are optional.
- Medium Zone: Hazards with moderate severity but infrequent/intermediate probability of occurring are conditionally acceptable hazards requiring further effects analysis to create mitigation plans to control risks.
- High Zone: Hazards with critical to catastrophic severity and a frequent probability of occurring are unacceptable and require a risk management procedure to mitigate further so that it falls into an acceptable risk zone.
Determining the level of risk for any given hazard sets the foundation for the overall risk management process. Note that no medical device industry standard exists for severity and probability of occurrence. You may need more than three levels of risk when performing your evaluation. The levels of risk you define should align with your medical devices.
Referring back to the pacemaker example, the risk of biohazard contamination is in the medium zone because it could be moderate in severity, with infections leading to the need for further medical care and treatment, but likely infrequent/average probability of occurring due to the precautions healthcare providers take before surgery. The possibility of electrical failure is likely in the high zone for this device because it only has one battery/power source, and it’s common for batteries to run out of power. The severity is catastrophic in this example because the malfunction of a pacemaker will lead to heart arrhythmia and potentially death. Both hazards require mitigation plans for risk management
Are you seeking guidance on evaluating risk for your medical device? Contact our team. We’ll help you define the levels of risk and perform the evaluation.
Step 3: Manage Risk with a Mitigation Plan
Once hazards with needs for risk management are identified, you can then develop a mitigation plan. According to the ISO/TR 24971:2020, all risks must be reduced “as far as possible.” Mitigations can be implemented to reduce a hazard’s severity or probability.
Generally, the first type of mitigation involves avoiding or eliminating the causes of the failure or hazard. If that isn’t possible, mitigation plans may include methods of detecting failures or hazards earlier to reduce the severity of the outcome. Finally, mitigation may involve reducing the impact of hazards/failures in medical devices.
Here are some risk management tactics:
- Incorporate human factors early in the process. Considering human factors and usability engineering in the initial design of a device ensures device user interfaces are designed to reduce use errors. Not only can this avoid risk and future (potentially costly) design changes, but it also aligns with the FDA’s guidance and IEC 62366-1:2015.
- Alter the designs of medical devices. The design of medical devices can be modified to reduce risk. Design changes may include altering a device’s shape, size, material, or other physical characteristics to improve safety and effectiveness. It’s important to note that altering the design can lead to further risk management concerns and require careful attention.
- Include risk management measures during manufacturing that decrease the frequency of harm. This approach is effective in upholding the original intent while promoting safety.
- Include clear instructions. While this may seem intuitive, it is necessary to mention it. Thoughtfully constructed instructions on how to use the medical device, highlighting potential hazards and providing effective preventive measures, can help reduce the risk of assumption, misinterpretation, and misuse.
In the pacemaker example, there is a clear need for risk management and mitigation plans. To reduce the risks associated with biohazard contamination, healthcare providers may be warned with clear instructions to suggest sterile handling and implantation. Furthermore, a medical device manufacturer may implement risk management activities during the manufacturing, including sterilization techniques to avoid biohazard contamination. Finally, the possibility of electrical failure can be mitigated by altering the device’s design to include a redundant power system/battery so that if one battery/power system fails, the second one will avoid malfunction.
11 Terms in ISO 14971:2019 to Master
Three new terms, ‘benefit,’ ‘reasonably foreseeable misuse,’ and ‘state of the art,’ were specified in ISO 14971:2019. These terms were frequently used in the older versions of ISO 14971. However, the 2019 version has added definitions and explanatory notes to clarify their meanings. It enables the readers to understand the concept behind the terms with ease. Below we list vital terms and definitions to remain familiar with.
- Benefit: Positive impact or desirable outcome of using a medical device on an individual’s health or a positive effect on patient management or public health.
- Note: Benefits can include a positive impact on clinical outcomes, the patient’s quality of life, outcomes related to diagnosis, a positive impact from diagnostic devices on clinical outcomes, or a positive impact on public health.
- Harm: Injury or damage to the health of people or damage to property or the environment.
- Hazard: Potential source of harm.
- Hazardous situation: Circumstance in which people, property, or the environment is/are exposed to one or more hazards.
- Intended use: Use for which a product, process, or service is intended according to the specifications, instructions, and information provided by the manufacturer.
- Note: The intended medical indication, patient population, part of the body or type of tissue interacted with, user profile, use environment, and operating principle are typical elements of the intended use.
- Reasonably foreseeable misuse: Use of a product or system in a way not intended by the manufacturer but can result from readily predictable human behavior.
- Note: Readily predictable human behavior includes all types of users, e.g., lay and professional users. Reasonably foreseeable misuse can be intentional or unintentional.
- Residual risk: Any risk that remains even after various risk control and mitigation measures have been implemented to reduce potential risks to an acceptable level.
- Risk: Combination of the probability of occurrence of harm and the severity of that harm.
- Risk control: Making decisions and implementing measures to mitigate risks to specified levels.
- Safety: Freedom from unacceptable risk.
- State-of-the-art: Developed stage of technical capability at a given time regarding products, processes, and services based on the relevant consolidated findings of science, technology, and experience.
- Note: The state-of-the-art embodies what is currently and generally accepted as good practice in technology and medicine. State-of-the-art does not necessarily imply the most technologically advanced solution. The term “state-of-the-art” mentioned here is occasionally called the “generally accepted state-of-the-art.”
The Latest Updates with ISO 14971 Risk Management
Risk Management for Devices Sold in Europe
As mentioned earlier, if selling devices in Europe, a manufacturer must consider BS EN ISO 14971:2012 from the British Standards Institute (BSI), approved by the European Committee for Standardization (CEN) on May 16, 2012. Its content is identical to ISO 14971:2007; however, three annexes at the beginning identify several shortcomings within ISO 14971:2007 compared to the Medical Device Directive (MDD) 93/42/EEC.
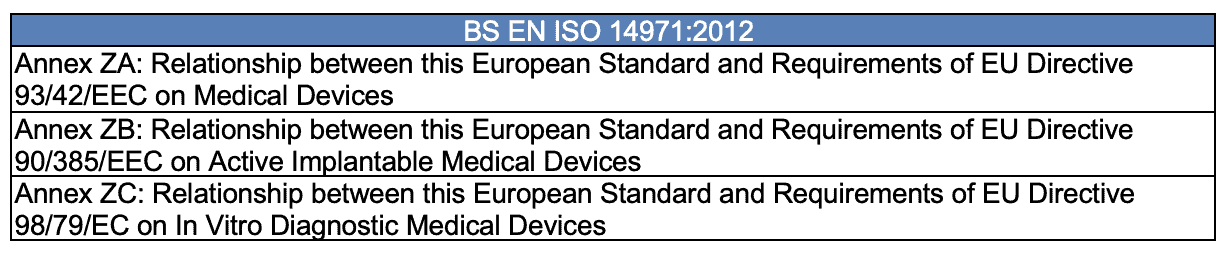
The standard text within these unique BS EN ISO 14971:2012 annexes is that all risks have to be reduced “as far as possible” vs. allowing the manufacturer to have the freedom to decide upon the threshold for risk acceptability.
Migration of the Annexes and Cybersecurity
Six annexes from ISO 14971:2007 were moved to the guidance on the application of ISO 14971, and two were added, resulting in a total of eight annexes now in ISO/TR 24971:2020.

@ – moved from ISO 14971:2007
Shifting and expanding the annexes from the standard to the guidance will allow for more frequent revisions. This change highlights the revision’s emphasis on addressing the evolving needs of medical device software.
Flexibility to update the guidance will enable our industry to focus more aggressively on medical device cybersecurity and patient privacy issues as the technology changes far faster than the standard, which is reviewed every five years.
As our industry expands to include more medical devices with a software component, we must ensure that the application of standards that ensure safety can keep pace with the speed of technology – the most recent updates to the standards and their guidance are a step in the right direction.
Manage Risk with a Trusted Partner
Overall, risk management is a process that helps organizations identify and assess potential risks. Through hazard identification, risk evaluation, and mitigation planning, medical device companies can ensure they take all necessary precautions to keep patients safe from harm.
When creating a medical device risk management plan, consider hiring experts with the experience and expertise to assist. Ideal partners will offer guidance on identifying and mitigating hazards. They will understand the regulations and standards applicable to medical devices, which can help ensure your plan complies with those requirements. Finally, their knowledge and understanding of best practices for risk management plans may help you create a sound strategy for managing any potential issues surrounding your medical devices. By involving experts in your risk management plan, you can add a layer of safety and assurance that your product meets all industry standards and is safe for patients.
Contact QA Consulting today to discuss your medical device risk management strategy.