
Author: Amber Hilfiger, Director of Operations and Senior Engineering Consultant
A biological evaluation report (BER) is a collective summary of data used to demonstrate how compatible a medical device is with the human body. In other words, the BER provides evidence of a medical device’s biological safety, known as biocompatibility.
Medical device manufacturers must be aware of the various biocompatibility considerations during the development phase of the project. Because a device’s biocompatibility is assessed based upon its final, finished form, each process and material in contact with the device during its manufacture, including sterilization and packaging materials, should be considered in order to make informed choices.
The level of patient contact and interaction with the device determine the biological endpoints, or testing, that is required to demonstrate biocompatibility. At a minimum, every device that has some patient contact, whether direct or indirect, must consider the biological endpoints of cytotoxicity, sensitization, and irritation. These three main tests are known in the industry as the “Big 3.”
To legally market your device, it must be ISO 10993-1:2018 compliant. Specific to marketing your product in the U.S., the FDA provides guidance on using the standard.
Continue reading to learn:
- Details about the history and spirit of ISO 10993-1:2018
- Hallmarks of an exemplary BER
- Three common mistakes to avoid
- Consequences of a poorly written BER
ISO 10993 and Endpoint Testing
ISO 10993 was initially released nearly 30 years ago to help medical device manufacturers select the appropriate biocompatibility test for their devices. However, the idea of a risk management process was not added to the standard until the 2009 revision.
As a general rule of thumb, the required biological endpoint data for a device, acquired via testing, is determined by the nature of contact, the duration of contact, and the body systems involved in the device contact (e.g. intact skin, mucosal tissue, circulating blood, bone). This information is navigated via a biocompatibility evaluation matrix provided by ISO 10993-1 and further supported by FDA guidance: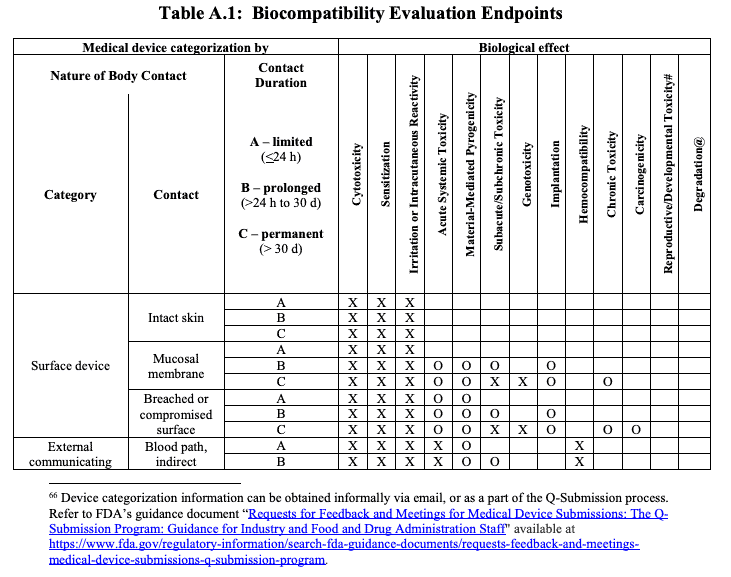
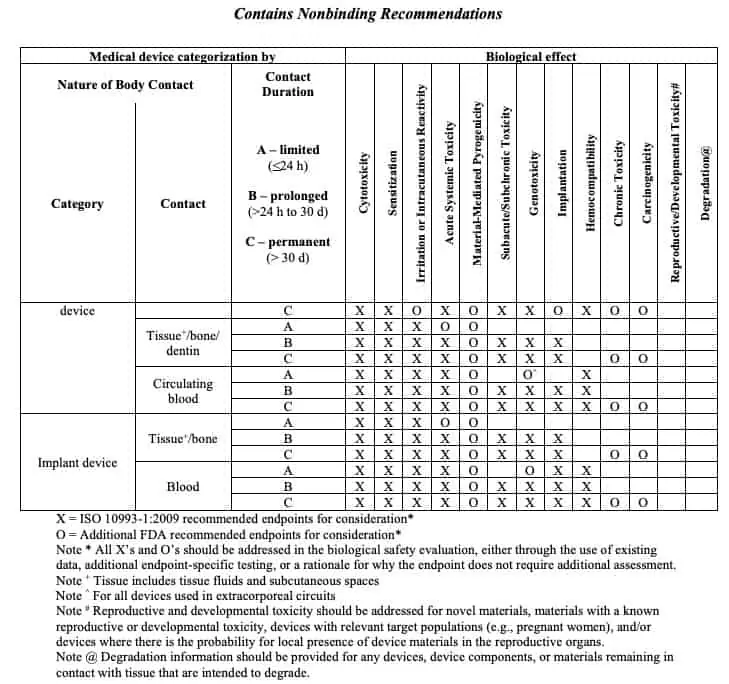
There are circumstances in which a rationale may be accepted in place of biological endpoint testing. Devices manufactured from materials that have been well characterized both chemically and physically and/or have a long history of safe use in marketed devices may support a rationale that the biocompatibility of a device has been established. This is particularly the case where a rationale based upon existing data may minimize the necessity of animal testing. Note that the risk profile of a device will also determine how much existing data may support a biocompatibility rationale in lieu of biological endpoint testing.
Tips for Writing an Exemplary BER
Based on my experience in the industry, here are four tips for writing an exemplary BER:
- Start early. Manufacturers should be early enough in the development process that the BER is a living document used to map gaps and identify where the biological safety of your device may be acceptable but could be improved.
- Identify the risk first. Assess and identify the risks associated with your device BEFORE you start the evaluation process. Document the Instructions for Use (IFUs), schematics, intended use, targeted population, maximum exposure, and other relevant details.
- Don’t go it alone. Partner with experts who have the technical competencies and training to interpret ISO 10993-1 and the FDA guidance, and who know how to correctly apply them to the evaluation of your device.
- Revisit your BER. Even after your device is determined to be biologically safe, manufacturers should revisit the BER annually to determine if there are opportunities for increased safety, or if changes have occurred that may impact the safety of the product.
3 Common Mistakes to Avoid
Here are three of the most common mistakes our team encounters in helping clients with their BERs:
- Poor data collection
- Failure to consider all the materials of a device that have patient contact
- Selection of materials by price (without considering the cost of testing)
Don’t Overlook Risk Assessment
One of the most overlooked elements of a pragmatic biological evaluation report is a thorough risk assessment.
The more extensive and invasive the patient contact with the device, the higher the risk associated with the medical device. It’s critical that you document a structure and hierarchy of risk and decide each level’s evaluation criteria.
Consequences of a Weak Biological Evaluation Report
A poorly written biological evaluation report can delay the clearance or approval to market your medical device and have other downstream consequences.
First, because you did not present a strong enough argument to regulators, they may enforce lengthy and expensive testing, leading to further delays and increased costs.
Second, you are wasting time not testing. If the existing data doesn’t support not testing, you’re simply extending the timeline instead of using your time efficiently to complete necessary tests.
Lean into our Microbiology Know-How
With experienced microbiologists on staff, the QA Consulting team is well-versed in understanding the biocompatibility and endpoint testing required for all classes of medical devices.
Lean into our medical device microbiology know-how to avoid common pitfalls and the downstream consequences of a poor BER.
Contact us today to talk to a medical device quality consulting expert about microbiology analysis and let us develop your next biological evaluation report.