
FDA Inspection Observations (483s) — Current Trends and Preventing Escalation
Ideally, you will have tactics and strategies in place to avoid receiving FDA Form 483s and preventing problems from escalating. Here, we shed light on the recent trends in FDA inspection observations, changes due to the pandemic, and tactics and strategies for mitigating escalation.
A quick recap on FDA 483s:
An FDA Form 483 is issued after an inspection when an investigator has observed conditions that violate the Food Drug and Cosmetic (FD&C) Act. Medical device companies are responsible for taking corrective action to address the cited objectionable conditions and any related non-cited objectionable conditions that might exist.
We help companies respond to the FDA Form 483 while developing and implementing prompt corrective action plans.
FDA 483 Trends in Recent Years
Based on the FDA’s 483 database system, a total of 3,838 483s were issued from October 2021 through September 2022—with 547 issued for Devices and Radiologic Health.
Main issues driving 483 observations (based on data samples from 2020, 2021, and 2022):
- Design Controls
- CAPAs
- Complaints
- Management responsibility
- Medical Device Reporting
The ranking of each issue varies depending on the source of information. However, the issues listed above rank highly across data sources.
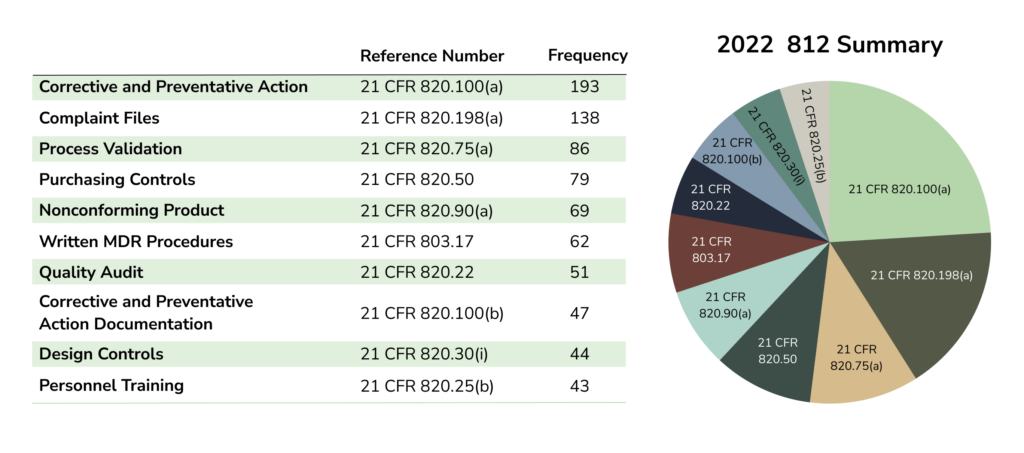
483 Analysis of 2022 Published 483s
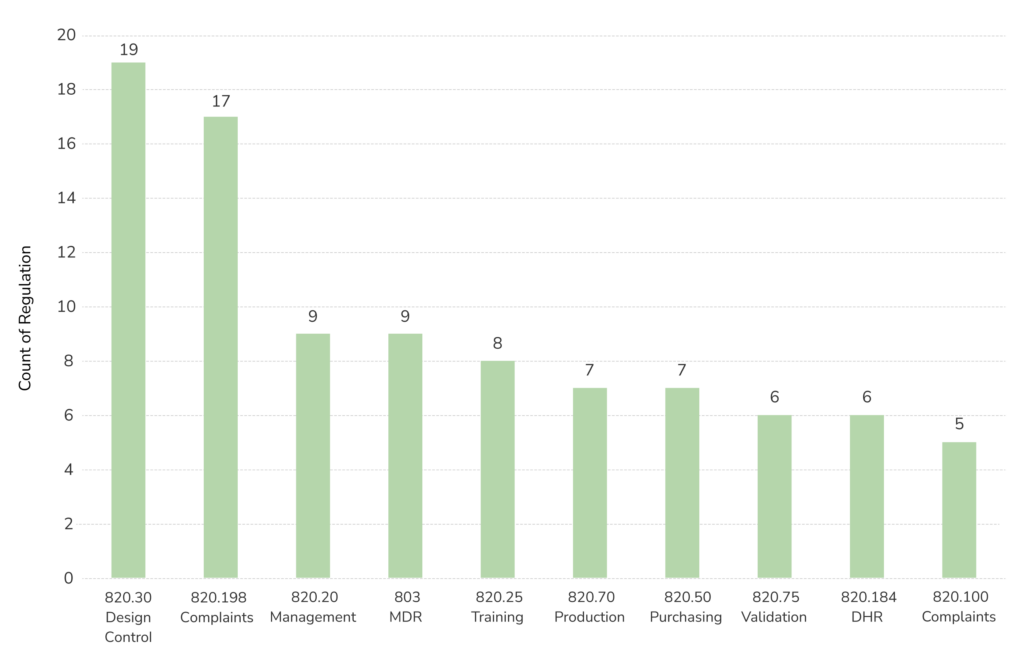
When looking at devices and radiological health programs alone, 483s issued dramatically decreased from the previous year, with 431 issued from October 2019 through September 2020. This is partly due to changes associated with the pandemic.
The Pandemic and Mission-Critical Activities
While there are multiple types of inspections, the gold standard has always been onsite inspections. These inspections provide a snapshot of the company and its operations. Due to the pandemic, routine on-site FDA inspections were put on hold in March 2020. They have now resumed based on risk factors to health and safety.
To see the effect of the pandemic on the FDA’s inspectional activities, check out the Resiliency Roadmap for FDA Inspectional Oversight. You’ll also find the FDA’s priorities moving forward and their plan for establishing consistency in their operations.
The FDA reserves inspections for mission-critical issues. Therefore, the Resiliency Roadmap emphasizes mission-critical areas.
Factors Determining Mission-Critical Inspections:
- Product received breakthrough therapy or regenerative medicine advanced therapy designation
- Product is used to treat a serious disease or medical condition, and there is no substitute
- Product requires follow-up due to recall, or there is evidence or serious adverse events or outbreaks of a foodborne illness
- Product is related to FDA’s COVID-19 response (e.g., drug shortage)
Mission-critical work can go beyond inspections to sample collections, analyses, recall activities (such as recall audit checks) and more. It is vital to remain aware of mission-critical activities, how they are enforced, and how you can prepare.
QA Consulting can help you remain aware and prepared.
FDA’s Approach to Optimizing Surveillance
FDA inspections have resumed through the FDA’s Office of Regulatory Affairs (ORA) with the sole purpose of monitoring and enforcement. However, recent inspection trends have influenced the number of Form 483s issued.
Inspection Trends:
- Increase in “For Cause Inspections”
- Virtual inspections were substituted for on-site inspections during Covid
- Foreign inspections stopped
How can you prepare?
Anticipation of objectionable FDA-regulated product requirements can help prevent the potential escalation of problems.
Expert Insights:
- A FDA 483 letter only begins the process of investigating objectionable conditions.
- FDA medical device warning letters can be avoided, particularly if there is a heightened awareness of related non-cited objectionable conditions.
- The 483 response must be comprehensive and consider other devices in addition to the observation. A system-level approach is a must.
Preventing Escalation: Tactics vs. Strategy
The best way to prevent escalation of problems arising from FDA 483 observations is not tactics or strategies. It’s both.
Tactics are concrete, easy-to-implement initiatives used to reach short-term goals. The most effective method of preventing an FDA 483 observation from escalating to an FDA warning letter is to take action, such as CAPA, on time.
However, tactics are not always enough.
Strategies are considered the long-term vision and must be created with care.
Tactics are aimless without an aligned strategy. If it remains unclear how an immediate action fits into a long-term strategy, consider whether the tactic should be part of it.
The most effective decisions combine good strategy with proven actions. They are data-informed with clearly defined goals. To de-risk your strategy, establish contingency plans or “what-ifs” as early as possible.
Tactical performance and strategic reasoning are especially relevant as we emerge from the COVID pandemic. The key to surviving FDA 483s and warning letters is awareness of current trends, marketplace changes, and regulatory initiatives.
We can help you define your goals, develop tactics and strategies, and remain aware of current trends.
FDA Inspection Tactics
Developing a standard operating procedure (SOP) is the first actionable step in the process of managing an FDA inspection. A good SOP will clearly state:
- Whom to contact first when the inspector shows up.
- How you will notify the entire facility that an inspector is in the building.
- How you will document FDA requests for information.
- How you will handle the inspector’s request for photographs or videos.
- How you will respond to an FDA request that is not in agreement with your quality procedures.
- How to define team members and train all relevant personnel to the SOP including subject matter experts (SMEs).
Remember — respecting boundaries and proper communication during the FDA inspection process is critical.
FDA Inspection Strategies
The components of an effective FDA inspection strategy are complex and multi-layered. The overall strategy can be divided into the following categories:
Quality Systems Compliance
Utilizing a comprehensive Quality Management System (QMS) will provide the foundation for an efficient and compliant company structure. The best QMS will be divided into a logical series of well-designed SOPs using a multi-layered approach.
QA Consulting can provide the necessary tools and expertise in QMS development and implementation.
QA Consulting maintains a spotless track record. None of the companies QA Consulting has worked with have been issued a Warning Letter or Major Nonconformance.
Internal or mock FDA audits
An essential element of success lies in preparation. Using the expertise of an outside consultant, such as QA Consulting, helps you prepare to conduct internal and mock audits. Internal audits take place within the company to ensure all procedures are compliant. Mock audits provide valuable information in a low-stress practice environment.
Internal compliance audits are required before the EU notified body audit. QA Consulting’s auditors are all certified in Quality Management System auditing and can guide medical device companies through internal compliance audits.
FDA 483 remediation
The strategy in post-market compliance demands knowledge and proficiency. QA consulting can assist with a wide range of post-market activities.
An FDA form 483 will list issues found during the audit. Despite best efforts, many companies will encounter at least one Form 483 at some point after FDA inspections. Remediation should begin immediately and in a logical, careful manner.
Don’t do it alone. QA Consulting can guide your company through an expert response method that walks you through a 483 response without escalating to a Warning Letter.
A response to an FDA inspection should be grounded in a foundation of quality subsystems, including:
- Corrective and preventive actions (CAPA)
- Design controls
- Management controls
- Production and process controls (P&PC)
A comprehensive response to an FDA 483 should ensure compliance, mitigate risks, and focus on as few disruptions as possible to medical device manufacturing workflow.
FDA Inspection Expertise
Most FDA 483 observations and FDA warning letters in 2022 were the result of onsite inspections, replacing pandemic-era trends. The shift away from alternative tools for enforcement is likely to continue with a risk-based approach. Therefore, an increase in “For Cause” Inspections, particularly in high-risk areas, will continue.
Tactical performance and strategic reasoning are not mutually exclusive. Developing the right processes to deal with FDA Inspection Methods and the potential results must align with a company’s core values.
QA Consulting assists companies in creating and implementing a long-term vision for success.